The Elastic High Performance Computing (E-HPC) client provides a graphical interface that simplifies the configurations of software applications such as GROMACS. You can use the client to perform a molecular dynamics simulation.
Background information
GROningen MAchine for Chemical Simulations (GROMACS) is a full-featured software package. It is used to perform molecular dynamics simulations based on Newtonian equations of motion for systems that include millions of particles. GROMACS is used for nucleic acid analysis of biochemical molecules such as proteins and lipids that have various complex bonded interactions.
This topic uses GROMACS as an example to describe how to use an E-HPC client to run an application. The procedure contains the following operations:
Connect to a cluster by using the terminal of an E-HPC client. Download and decompress a file in the cluster.
Configure GROMACS and submit a job.
Query the result of the job.
Connect to a cloud desktop by using Virtual Network Computing (VNC). Use VMD to view the result of the job.
Preparations
Before you use the E-HPC client to submit a job, make sure that you have completed the following preparations for your cluster:
The following software is installed in the cluster. For more information, see Install software:
VMD V1.9.3
openmpi V3.0.0
GROMACS is installed on each compute node of the cluster.
Sample command:
sudo yum install -y gromacs sudo cp /usr/bin/gmx /usr/bin/gmx_mpitmux is installed. If the cluster is deployed in Tiny mode, install tmux on the management node. If the cluster is deployed in Standard mode, install tmux on the logon node.
Sample command:
sudo yum install -y tmuxVNC is enabled for the cluster. For more information, see Use VNC to manage a visualization service.
A rule is configured to enable the following ports for the security group to which the cluster belongs:
12011: Port 12011 is used to log on to the cluster.
1201x starting from 12017: Port 12017 allows only one user to open the VNC Viewer window. If multiple users need to open the VNC Viewer window, you need to enable the corresponding number of ports, starting from port 12017. For example, if three users need to use the VNC service, you need to enable ports 12017, 12018, and 12019.
For more information, see Add a security group rule.
Procedure
Log on to an E-HPC client.
For more information, see Log on to an E-HPC client.
Connect to the cluster by using the terminal of the E-HPC client. Download and decompress a file in the cluster.
In the left-side navigation pane, click Session Management. On the Session Management page, click terminal to open the Terminal window.
Run the following command to download and decompress a file.
This example simulates water molecules to track the motion of water molecules in a specified space at a specified temperature.
wget https://public-ehpc-package.oss-cn-hangzhou.aliyuncs.com/water_GMX50_bare.tar.gz tar xzvf water_GMX50_bare.tar.gzUse an SSH tool to switch to the compute nodes. Run the following command to preprocess the file:
cd water-cut1.0_GMX50_bare/0012/ gmx_mpi grompp -f pme.mdp -c conf.gro -p topol.top -o topol_pme.tpr
Check whether the VNC service is available.
In the upper-right corner of the Session Management page, click VNC to test whether the VNC service is available.
If the VNC service is connected, it is available.
Otherwise, check whether the VNC service is installed and whether the ports required by the VNC service are enabled.
Configure GROMACS and submit a job.
In the left-side navigation pane, click Application Center.
On the Application Center page, click gromacs.
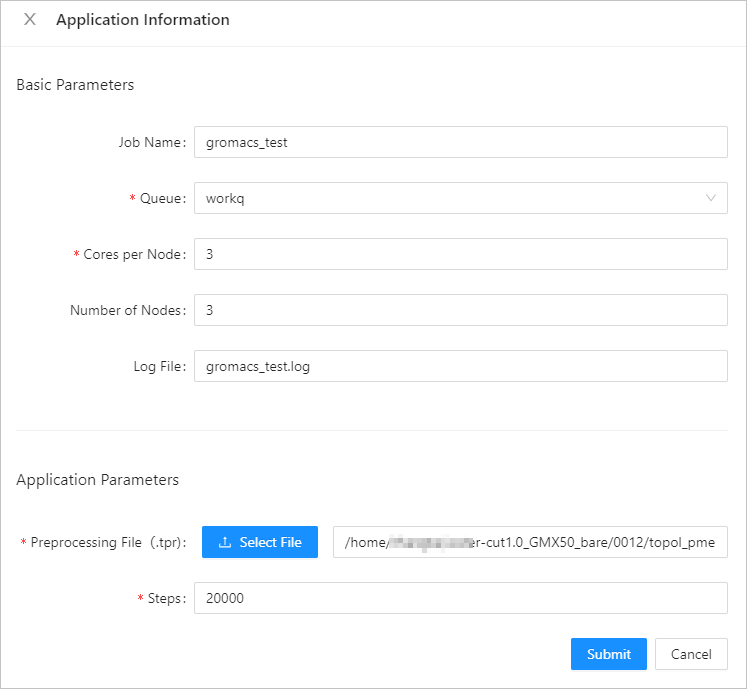
In the panel that appears, configure the parameters of GROMACS and click Submit.The following table describes the parameters.
Section
Parameter
Example
Description
Basic Parameters
Job Name
gromacstest
The name of the job.
Queue
workq
The queue that you want to run the job.
Number of Cores
16
The number of CPU cores of a single node.
Number of Nodes
3
The number of compute nodes that are required to run the job.
Log Output
gromacs.log
The output path of logs that are generated for the job.
Application Parameters
Preprocessed TPR File
/home/user***/water-cut1.0_GMX50_bare/0012/topol_pme.tpr
Upload the preprocessed file.
Maximum Number of Iterations
1000
The maximum number of iterations for the simulation. In the file of the example, the nsteps parameter specifies the maximum number of iterations.
Query the result of the job.
In the left-side navigation pane, click Job List.
Configure query conditions to query the result of the job.
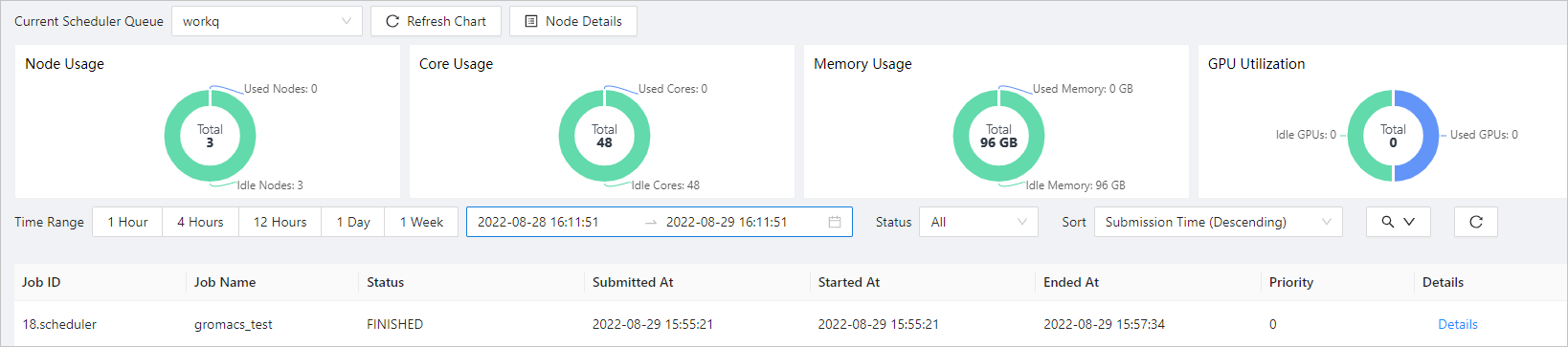
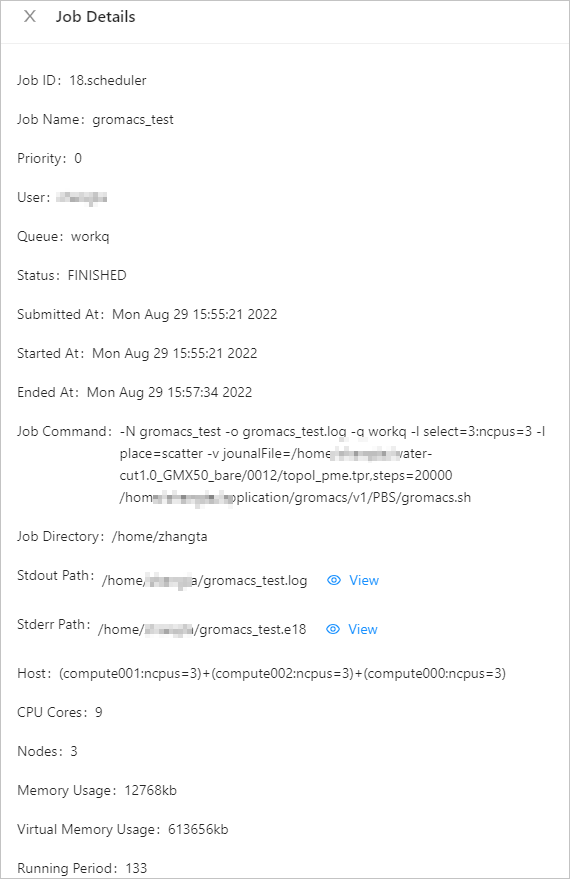
If the job enters the FINISHED state, the job is completed. Click Details to view the job details.
After the job is completed, configure VNC to view the job result.
In the left-side navigation pane, click Application Center.
On the Application Center page, click gromacs-vnc.
In the panel that appears, select the file that you want to view and click Submit.
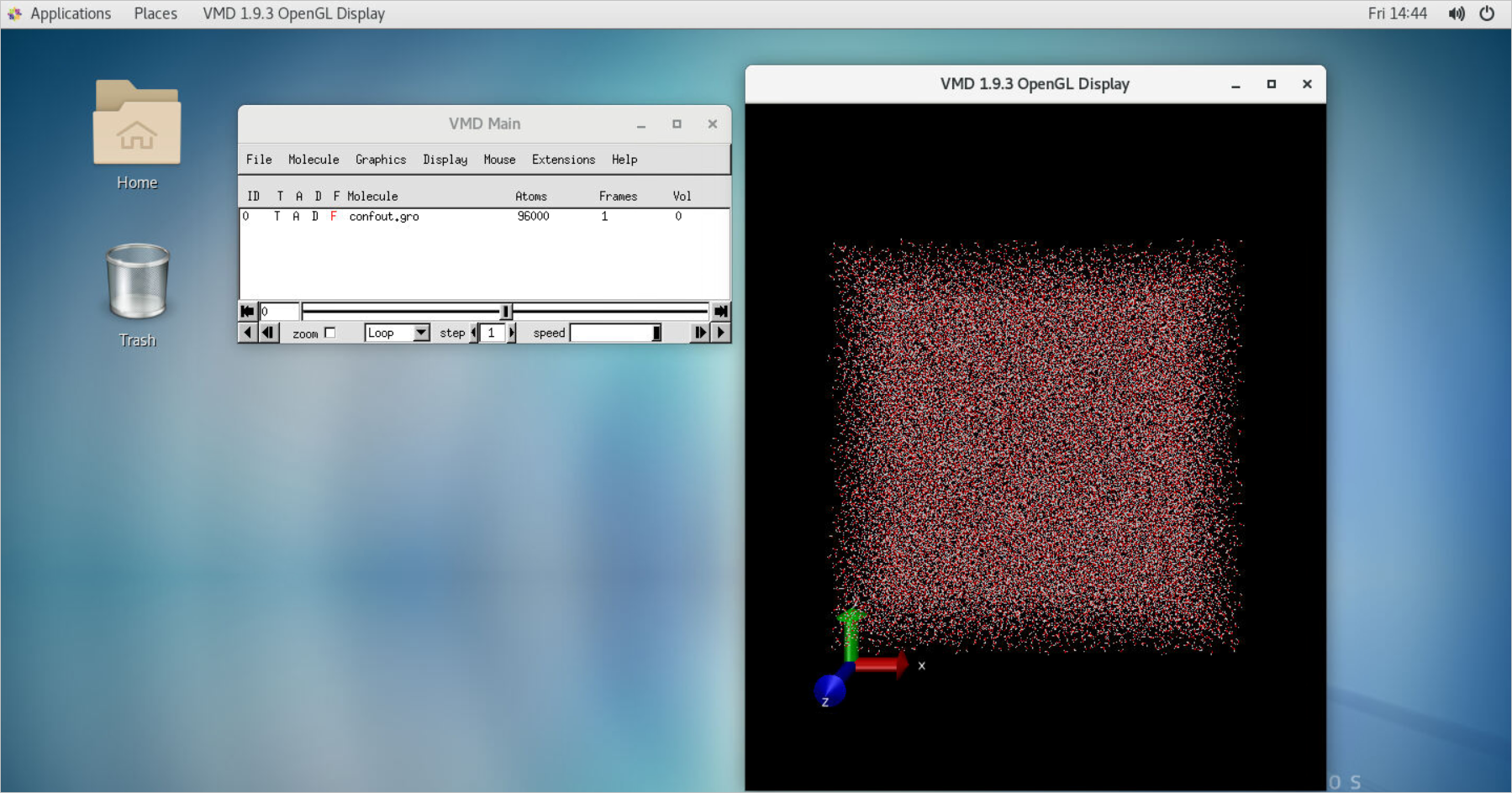
In the upper-right corner of the Session Management page, click VNC to open the VNC window. View the job result in the VMD window that appears.