This topic uses GROMACS as an example to describe how to perform a molecular dynamics simulation by using an Elastic High Performance Computing (E-HPC) cluster.
Background information
GROningen MAchine for Chemical Simulations (GROMACS) is a full software package. It is used to perform molecular dynamics by simulating Newtonian equations of motion for systems that include millions of particles.
GROMACS is used for nucleic acid analysis of biochemical molecules such as proteins and lipids that have various complex bonded interactions. GROMACS is the preferred method for computing typical simulations, for example, non-bonded interactions. Many researchers in different industries use GROMACS to study polymers in non-biological systems.
GROMACS supports common algorithms of molecular dynamics (MD). Graphics processing units (GPUs) can be used in GROMACS to accelerate critical computing tasks. For more information, visit the GROMACS official website.
Related examples
Example 1: lysozyme in water
In this example, GROMACS is used to simulate a protein that interacts with ions in a box of water.
To download the file of the example, click the following link: https://public-ehpc-package.oss-cn-hangzhou.aliyuncs.com/Lysozyme.tar.gz.
Example 2: motion of water molecules
This example is a simulation of water molecules. It is used to track the motion of water molecules in a specific space at a specified temperature.
To download the file of the example, click the following link: https://public-ehpc-package.oss-cn-hangzhou.aliyuncs.com/water_GMX50_bare.tar.gz.
Preparations
Create an E-HPC cluster. For more information, see Create a cluster by using the wizard.
The following parameter configurations are used in this example.
Parameter
Description
Hardware settings
Set the Deploy Mode to Standard. Specify two management nodes, one compute node, and one logon node. Select a GPU-accelerated instance type as the compute node. For example, you can select ecs.gn5-c8g1.2xlarge.
Software settings
Deploy a CentOS 7.2 public image and the PBS scheduler. Turn on VNC.
Create a cluster user. For more information, see Create a user.
The user is used to log on to the cluster, compile software, and submit jobs. The following settings are used in this example:
Username: gmx.test
User group: sudo permission group
Install software. For more information, see Install software.
Install the following software:
Gromacs-gpu V2018.1
openmpi V3.0.0
cuda-toolkit V 9.0
VMD V1.9.3
Step 1: Connect to the cluster
Connect to the cluster by using one of the following methods. This example usesgmx.test as the username. After you connect to the cluster, you are automatically logged on to the /home/gmx.test.
Use an E-HPC client to log on to a cluster
The scheduler of the cluster must be PBS. Make sure that you have downloaded and installed an E-HPC client and deployed the environment required for the client. For more information, see Deploy an environment for an E-HPC client.
Start and log on to your E-HPC client.
In the left-side navigation pane, click Session Management.
In the upper-right corner of the Session Management page, click terminal to open the Terminal window.
Use the E-HPC console to log on to a cluster
Log on to the E-HPC console.
In the upper-left corner of the top navigation bar, select a region.
In the left-side navigation pane, click Cluster.
On the Cluster page, find the cluster and click Connect.
In the Connect panel, enter a username and a password, and click Connect via SSH.
Step 2: Submit a job
Run the following command to download and decompress a file.
The example of the motion of water molecules is used in this topic.
wget https://public-ehpc-package.oss-cn-hangzhou.aliyuncs.com/water_GMX50_bare.tar.gz tar xzvf water_GMX50_bare.tar.gz chown -R gmx.test water-cut1.0_GMX50_bare chgrp -R users water-cut1.0_GMX50_bareRun the following command to create a job script file named gmx.pbs:
vim gmx.pbsThe sample script:
NoteIn this example, a job is submitted by the user named gmx.test. This job is run on a compute node that contains eight vCPUs and one P100 GPU card. You can adjust the configuration of compute nodes based on your computing requirements.
#!/bin/sh #PBS -j oe #PBS -l select=1:ncpus=8:mpiprocs=4 #PBS -q workq # Specify the environment variables on which the module command depends. export MODULEPATH=/opt/ehpcmodulefiles/ module load gromacs-gpu/2018.1 module load openmpi/3.0.0 module load cuda-toolkit/9.0 export OMP_NUM_THREADS=1 cd /home/gmx.test/water-cut1.0_GMX50_bare/0096 # Preprocess the file of the example to generate a TPR input file. /opt/gromacs-gpu/2018.1/bin/gmx_mpi grompp -f pme.mdp -c conf.gro -p topol.top -o topol_pme.tpr # -ntomp specifies the number of OpenMP threads associated with each process, and -nsteps specifies the number of iterations in the simulation. mpirun -np 4 /opt/gromacs-gpu/2018.1/bin/gmx_mpi mdrun -ntomp 1 -nsteps 100000 -pin on -s topol_pme.tprRun the following command to submit the job:
qsub gmx.pbsThe following command output is returned, which indicates that the generated job ID is 0.scheduler:
0.scheduler
Step 3: View the job result
Run the following command to view the job execution status.
qstat -x 0.schedulerThe following code provides an example of the expected returned output, where an
Rin theScolumn indicates that the job is running, anFin theScolumn indicates that the job is completed.Job id Name User Time Use S Queue ---------------- ---------------- ---------------- -------- - ----- 0.scheduler gmx.pbs gmx.test 00:34:42 F workqNoteWait until the job is completed. You can run the
cat gmx.pbs.o0to view the job output after the job is completed.Use VNC to view the result of the job.
Enable VNC.
NoteMake sure that the ports required by VNC are enabled for the security group to which the cluster belongs. When you use the console, the system automatically enables the port 12016. When you use the client, you need to enable the ports manually. Port 12017 allows only one user to open the VNC Viewer window. If multiple users need to open the VNC Viewer window, you need to enable the corresponding number of ports, starting from port 12017.
Use the client
In the left-side navigation pane, click Session Management.
In the upper-right corner of the Session Management page, click VNC to open VNC Viewer.
Use the console
In the left-side navigation pane of the E-HPC console, click Cluster.
On the Cluster page, select a cluster. Choose More > VNC.
Use VNC to remotely connect to a visualization service. For more information, see Use VNC to manage a visualization service.
In the Virtualization Service dialog box of the cloud desktop, choose Application > System Tools > Terminal.
Run the
/opt/vmd/1.9.3/vmdto open Visual Molecular Dynamics (VMD).On the VMD Main page, choose File > New Molecule....
Click the Browse... button and select the conf.gro file.
NoteThe path of the conf.gro file is /home/gmx.test/water-cut1.0_GMX50_bare/0096/conf.gro.
Click Load. You can view the result of the job in the VMD 1.9.3 OpenGL Display window.

Step 4: View the computing performance of the job
You can view the computing performance of a job only in the console.
Log on to the E-HPC console.
In the left-side navigation pane, choose Job and Performance Management > E-HPC Tune.
Find the cluster and click Node.
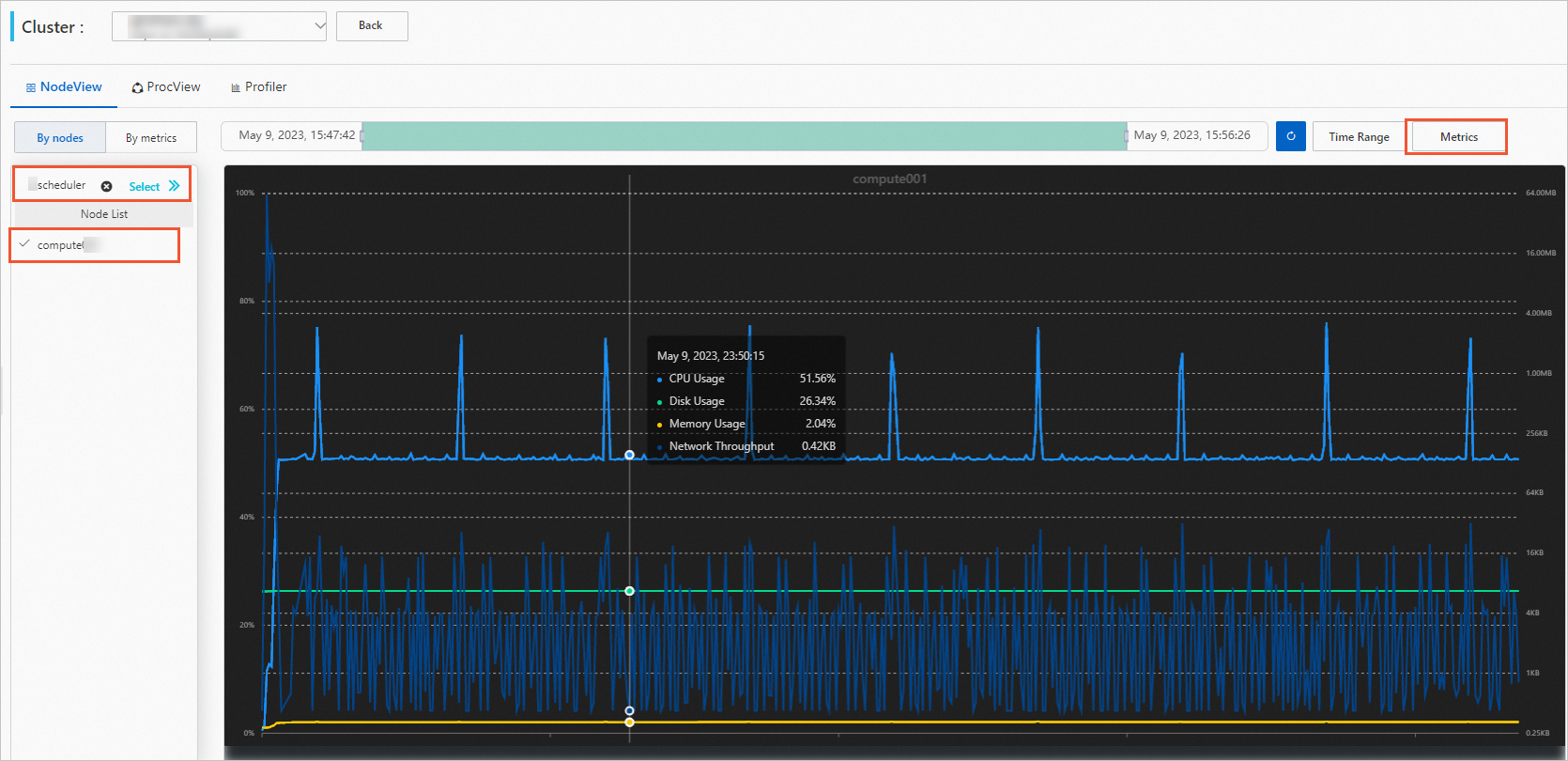
On the NodeView tab, view the performance of the node.
Select a job and a compute node.
Optional. Set a time period.
After you select a job, the time period is automatically set to the time period during which the job is run. You can also adjust the time period manually.
Click Metrics and select the metrics that you want to view.
View node performance data.
You can hover the mouse over the graph to view the detailed data.
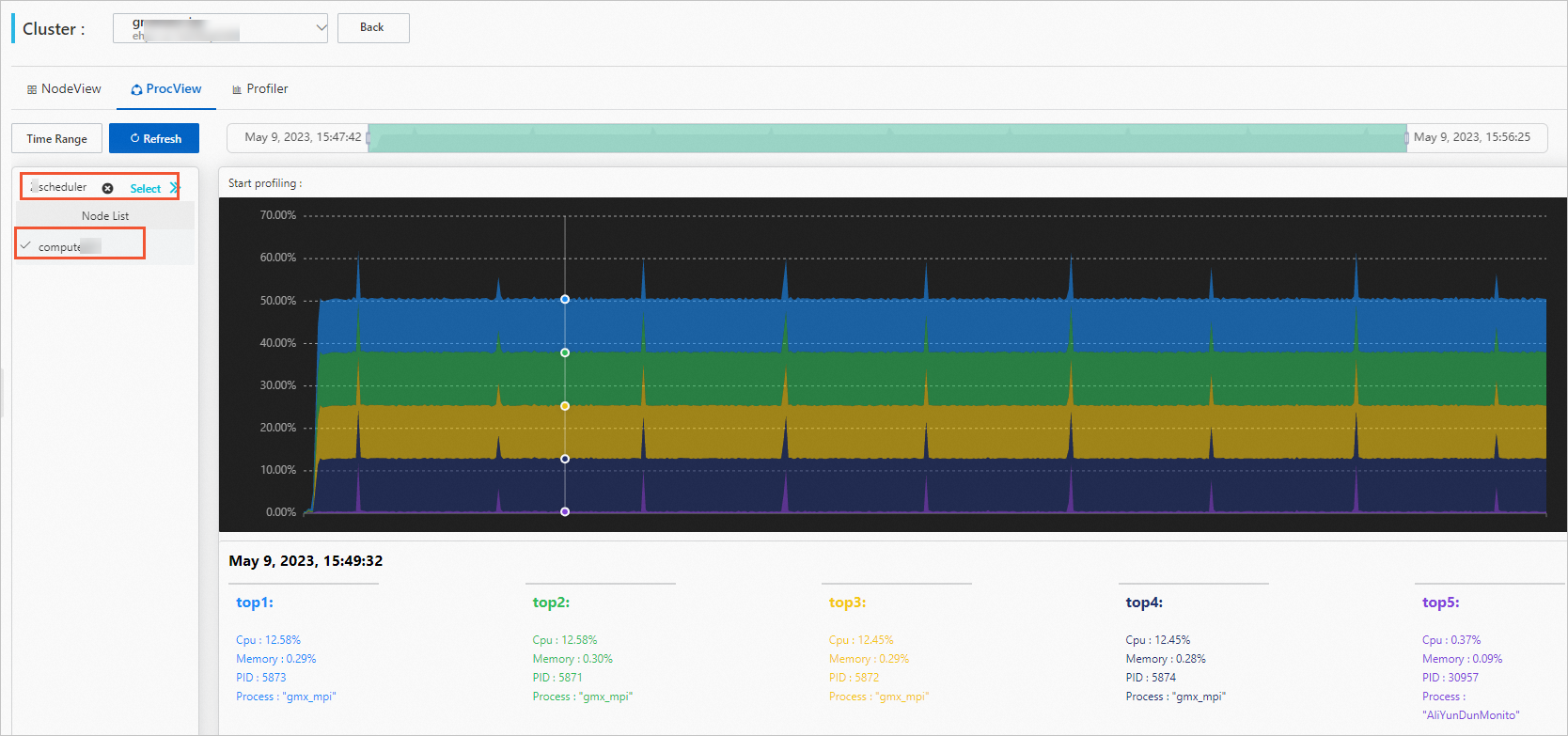
Click the ProcView tab to view the process performance.
Select a job and a compute node.
Optional. Set a time period.
After you select a job, the time period is automatically set to the time period during which the job is run. You can also adjust the time period manually.
View process performance data.
You can hover the mouse over the graph to view the detailed data.
Start a performance profiling task.
Click the point in time you want on the process performance graph, and then click the process you want to profile at the top of the graph.
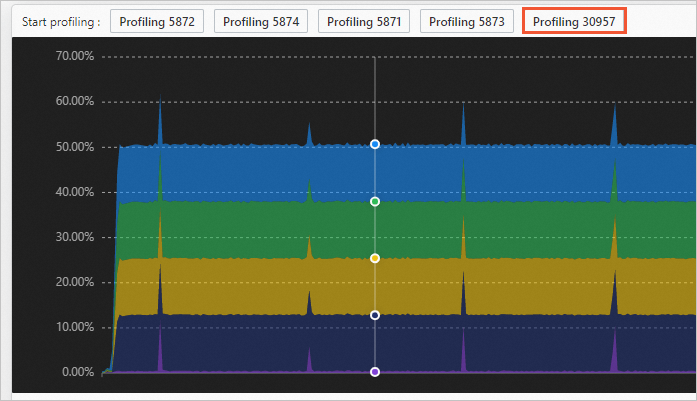
In the displayed dialog box, configure the profiling parameters.
Set a duration and frequency for the profiling and click OK to start real-time profiling for the job.
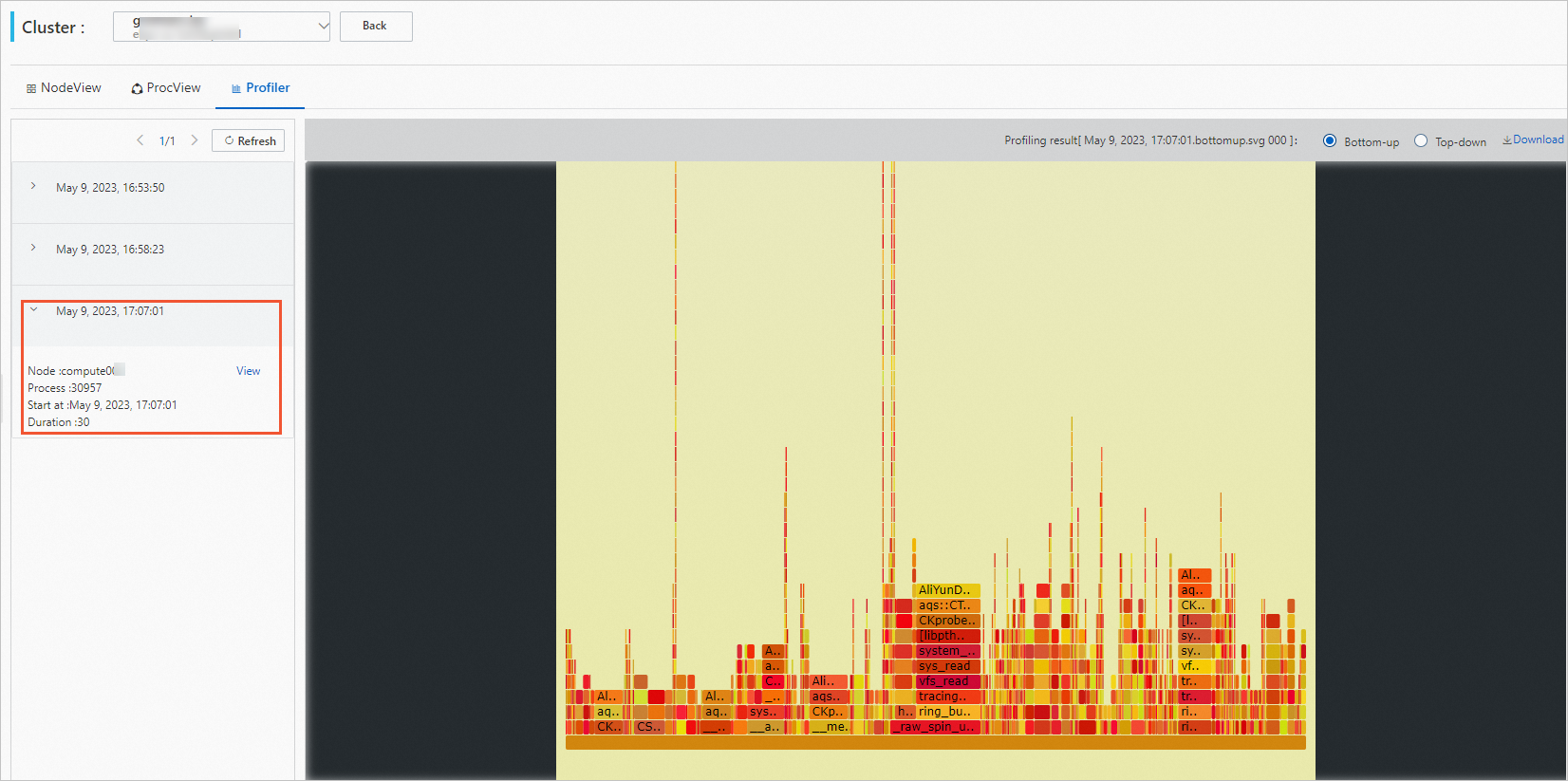
Click the Profiler tab to view the profiling result.
Select a profiling task and click View.
View the profiling results.
You can hover the mouse over the graph to view the detailed data.