このトピックでは、TairVector ベクトルエンジンを使用して、化合物や医薬品の分子構造の近似検索を実行するソリューションについて説明します。
背景情報
AI 創薬において、ベクトル検索は重要な役割を果たします。この分野では、ベクトルは化合物や医薬品を表します。ベクトル空間での類似度計算は、それらの相互作用を予測し、最適化するのに役立ちます。このメソッドにより、強い相互作用を持つ化合物や医薬品を迅速にスクリーニングでき、新薬開発を加速させます。ベクトル検索はまた、創薬スクリーニングの精度と効率を向上させ、医学研究者により効率的で正確な研究手法を提供します。
従来のベクトル検索サービスと比較して、TairVector はすべてのデータをメモリ内に格納し、リアルタイムのインデックス更新をサポートすることで、より低い読み書きレイテンシーを実現します。TVS.KNNSEARCH のような近傍探索クエリコマンドを使用すると、データベースから最も類似した上位 k 件の分子構造を効率的に取得できます。k の値はカスタマイズ可能です。これにより、人為的なミスや見落としによるプロジェクトの失敗リスクを低減します。
ソリューションの概要
以下のフローチャートにプロセスを示します。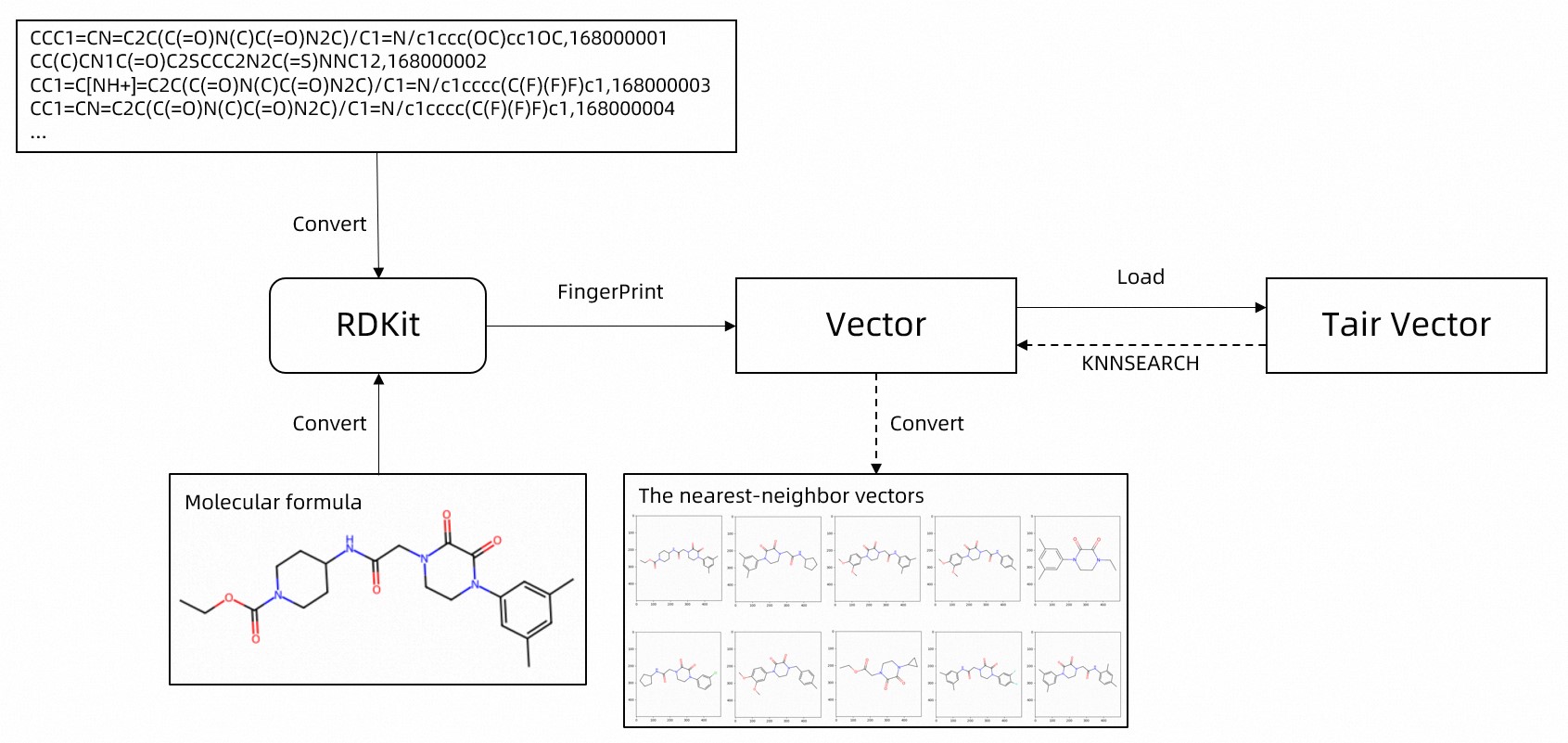
分子構造データセットをダウンロードします。
この例では、PubChem オープンソースデータセットのテストデータを使用します。これには 11,012 行のデータが含まれています。ダウンロードリンクのデータは、SMILES (Simplified Molecular Input Line Entry System) 形式です。次のサンプルは、化学式と一意の ID の 2 つの列を示しています。
説明実際のプロジェクトでは、より多くのデータを書き込んで、Tair のミリ秒レベルの検索性能をテストできます。
CCC1=CN=C2C(C(=O)N(C)C(=O)N2C)/C1=N/c1ccc(OC)cc1OC,168000001 CC(C)CN1C(=O)C2SCCC2N2C(=S)NNC12,168000002 CC1=C[NH+]=C2C(C(=O)N(C)C(=O)N2C)/C1=N/c1cccc(C(F)(F)F)c1,168000003 CC1=CN=C2C(C(=O)N(C)C(=O)N2C)/C1=N/c1cccc(C(F)(F)F)c1,168000004公式サイトからデータをダウンロードする場合、データは SDF (Structure-Data File) 形式です。次のコードを使用して SMILES 形式に変換する必要があります:
Tair インスタンスに接続します。詳細については、サンプルコードの
get_tair関数をご参照ください。Tair にベクトルインデックスを作成して、分子構造を格納します。詳細については、サンプルコードの
create_index関数をご参照ください。サンプルの分子構造データを書き込みます。詳細については、「サンプルコードの
do_load関数」をご参照ください。RDKit ライブラリを使用して分子構造データからベクトル特徴を抽出します。次に、TVS.HSET ベクトルエンジンコマンドを使用して、一意の ID、特徴情報、および化学式を Tair に格納します。
分子構造の類似検索を実行します。詳細については、「サンプルコードの
do_search関数」をご参照ください。RDKit ライブラリを使用してクエリ分子のベクトル特徴を抽出します。次に、TVS.KNNSEARCH ベクトルエンジンコマンドを使用して、Tair の指定されたインデックスで最も類似した分子構造をクエリします。
サンプルコード
この例では Python 3.8 を使用します。事前に次の依存ライブラリをインストールしてください:pip install numpy rdkit tair matplotlib。
import os
import sys
from tair import Tair
from tair.tairvector import DistanceMetric
from rdkit.Chem import Draw, AllChem
from rdkit import DataStructs, Chem
from rdkit import RDLogger
from concurrent.futures import ThreadPoolExecutor
RDLogger.DisableLog('rdApp.*')
def get_tair() -> Tair:
"""
Tair インスタンスに接続します。
* host:Tair インスタンスのエンドポイント。
* port:Tair インスタンスのポート番号。デフォルトは 6379 です。
* password:Tair インスタンスのデフォルトアカウントのパスワード。カスタムアカウントで接続するには、「'username:password'」の形式を使用します。
"""
tair: Tair = Tair(
host="r-bp1mlxv3xzv6kf****pd.redis.rds.aliyuncs.com",
port=6379,
db=0,
password="Da******3",
)
return tair
def create_index():
"""
分子構造を格納するためのベクトルインデックスを作成します:
* この例のインデックス名は "MOLSEARCH_TEST" です。
* ベクトル次元は 512 です。
* 距離メトリックは L2 です。
* インデックスアルゴリズムは HNSW です。
"""
ret = tair.tvs_get_index(INDEX_NAME)
if ret is None:
tair.tvs_create_index(INDEX_NAME, 512, distance_type=DistanceMetric.L2, index_type="HNSW")
print("create index done")
def do_load(file_path):
"""
分子構造データセットのパスを入力します。このメソッドは、分子構造のベクトル特徴を自動的に抽出し (smiles_to_vector)、データを TairVector に書き込みます。
このメソッドは、parallel_submit_lines、handle_line、smiles_to_vector、および insert_data 関数も呼び出します。
データは次の形式で Tair に格納されます:
* ベクトルインデックス名:"MOLSEARCH_TEST"
* キー:分子構造の一意の ID (例:"168000001")。
* 特徴情報:512 次元のベクトル情報。
* "smiles":化学式 (例:"CCC1=CN=C2C(C(=O)N(C)C(=O)N2C)/C1=N/c1ccc(OC)cc1OC")。
"""
num = 0
lines = []
with open(file_path, 'r') as f:
for line in f:
if line.find("smiles") >= 0:
continue
lines.append(line)
if len(lines) >= 10:
parallel_submit_lines(lines)
num += len(lines)
lines.clear()
if num % 10000 == 0:
print("load num", num)
if len(lines) > 0:
parallel_submit_lines(lines)
print("load done")
def parallel_submit_lines(lines):
"""
同時書き込みのスケジューリングメソッドです。
"""
with ThreadPoolExecutor(len(lines)) as t:
for line in lines:
t.submit(handle_line, line=line)
def handle_line(line):
"""
単一の分子構造の書き込みを処理します。
"""
if line.find("smiles") >= 0:
return
parts = line.strip().split(',')
try:
ids = parts[1]
smiles = parts[0]
vec = smiles_to_vector(smiles)
insert_data(ids, smiles, vec)
except Exception as result:
print(result)
def smiles_to_vector(smiles):
"""
分子構造のベクトル特徴を抽出し、SMILES 形式からベクトルに変換します。
"""
mols = Chem.MolFromSmiles(smiles)
fp = AllChem.GetMorganFingerprintAsBitVect(mols, 2, 512 * 8)
hex_fp = DataStructs.BitVectToFPSText(fp)
vec = list(bytearray.fromhex(hex_fp))
return vec
def insert_data(id, smiles, vector):
"""
分子構造のベクトルを TairVector に書き込みます。
"""
attr = {'smiles': smiles}
tair.tvs_hset(INDEX_NAME, id, vector, **attr)
def do_search(search_smiles,k):
"""
クエリ対象の分子構造を入力します。このメソッドは、Tair の指定されたインデックスにクエリを実行し、最も類似した k 個の分子構造を返します。
まず、クエリ構造のベクトル特徴を抽出します。次に、TVS.KNNSEARCH コマンドを使用して、最も近い k 個の分子構造の一意の ID を見つけます (この例では k=10)。最後に、TVS.HMGET コマンドを使用して、対応する化学式を取得します。
"""
vector = smiles_to_vector(search_smiles)
result = tair.tvs_knnsearch(INDEX_NAME, k, vector)
print("The 10 molecular structures most similar to the query target are as follows:")
for key, value in result:
similar_smiles = tair.tvs_hmget(INDEX_NAME, key, "smiles")
print(key, value, similar_smiles)
if __name__ == "__main__":
# Tair データベースに接続し、"MOLSEARCH_TEST" という名前の分子構造用のベクトルインデックスを作成します。
tair = get_tair()
INDEX_NAME = "MOLSEARCH_TEST"
create_index()
# サンプルデータを書き込みます。
do_load("D:\Test\Compound_168000001_168500000.smi")
# MOLSEARCH_TEST インデックスで、"CCOC(=O)N1CCC(NC(=O)CN2CCN(c3cc(C)cc(C)c3)C(=O)C2=O)CC1" に最も類似した 10 個の分子構造をクエリします。
do_search("CCOC(=O)N1CCC(NC(=O)CN2CCN(c3cc(C)cc(C)c3)C(=O)C2=O)CC1",10)次のサンプル出力は、コードが正常に実行されたことを示しています:
create index done
load num 10000
load done
The 10 molecular structures most similar to the query target are as follows:
b'168000009' 0.0 ['CCOC(=O)N1CCC(NC(=O)CN2CCN(c3cc(C)cc(C)c3)C(=O)C2=O)CC1']
b'168003114' 29534.0 ['Cc1cc(C)cc(N2CCN(CC(=O)NC3CCCC3)C(=O)C2=O)c1']
b'168000210' 60222.0 ['COc1ccc(N2CCN(CC(=O)Nc3cc(C)cc(C)c3)C(=O)C2=O)cc1OC']
b'168001000' 61123.0 ['COc1ccc(N2CCN(CC(=O)Nc3ccc(C)cc3)C(=O)C2=O)cc1OC']
b'168003038' 64524.0 ['CCN1CCN(c2cc(C)cc(C)c2)C(=O)C1=O']
b'168003095' 67591.0 ['O=C(CN1CCN(c2cccc(Cl)c2)C(=O)C1=O)NC1CCCC1']
b'168000396' 70376.0 ['COc1ccc(N2CCN(Cc3ccc(C)cc3)C(=O)C2=O)cc1OC']
b'168002227' 71121.0 ['CCOC(=O)CN1CCN(C2CC2)C(=O)C1=O']
b'168000441' 73197.0 ['Cc1cc(C)cc(NC(=O)CN2CCN(c3ccc(F)c(F)c3)C(=O)C2=O)c1']
b'168000561' 73269.0 ['Cc1cc(C)cc(N2CCN(CC(=O)Nc3ccc(C)cc3C)C(=O)C2=O)c1']結果
類似した分子構造を画像として描画することもできます。次の図に例を示します。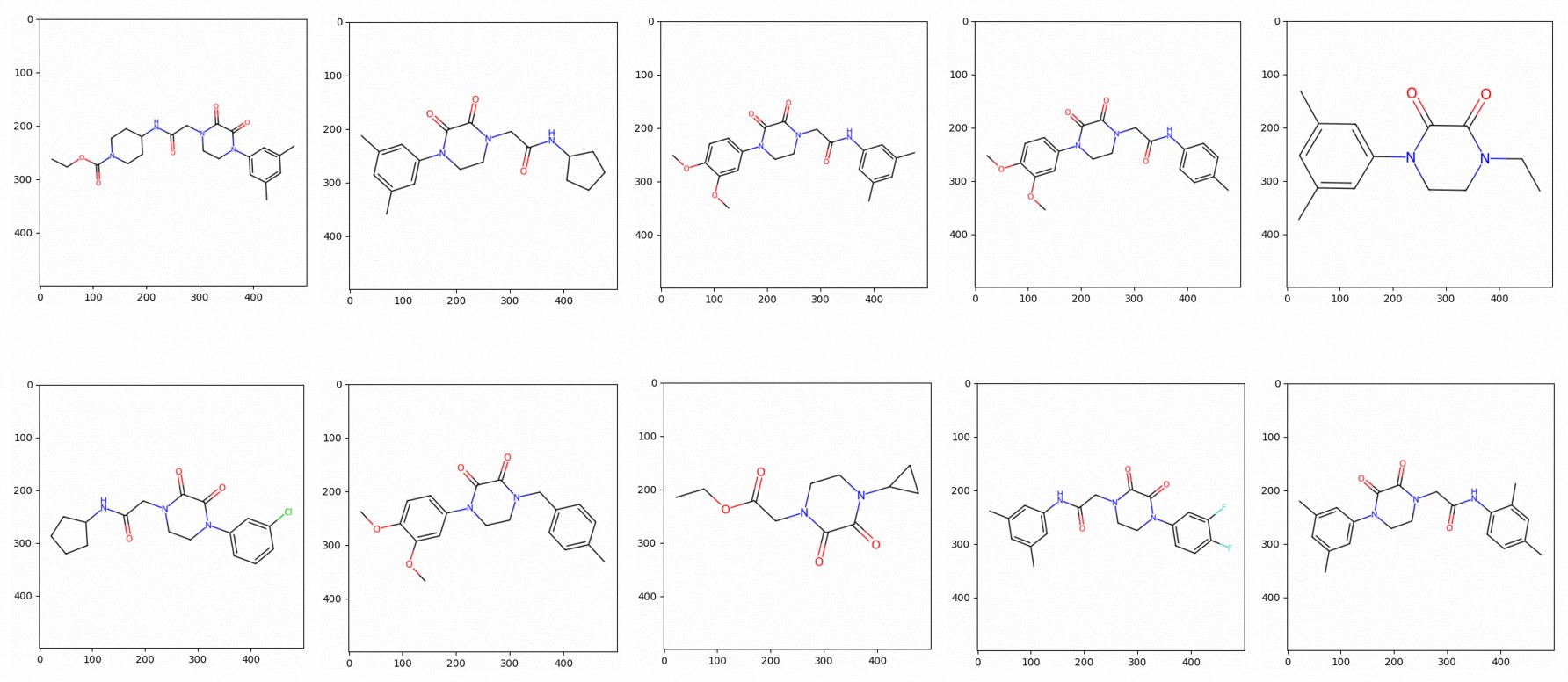
まとめ
TairVector を使用して分子構造を検索すると、最も類似した構造のリストをミリ秒単位で取得できます。Tair データベースにより多くの分子構造データセットが格納されるにつれて、後続のクエリはより正確かつタイムリーになります。このソリューションは、創薬分野における開発時間を短縮し、全体的な効率を向上させます。