This topic describes a solution that uses the TairVector vector engine to perform approximate searches for the molecular structures of compounds or drugs.
Background information
Vector retrieval plays a vital role in AI-powered drug discovery. In this field, vectors represent compounds and drugs. Similarity calculations in the vector space help predict and optimize their interactions. This method allows for the rapid screening of compounds and drugs with strong interactions, which accelerates new drug development. Vector retrieval also improves the accuracy and efficiency of drug screening, providing medical researchers with a more efficient and precise research method.
Compared to traditional vector retrieval services, TairVector stores all data in memory and supports real-time index updates for lower read and write latency. Nearest neighbor query commands, such as TVS.KNNSEARCH, allow you to efficiently retrieve the top k most similar molecular structures from the database. You can customize the value of k. This reduces the risk of project failures caused by human error or oversight.
Solution overview
The following flowchart shows the process.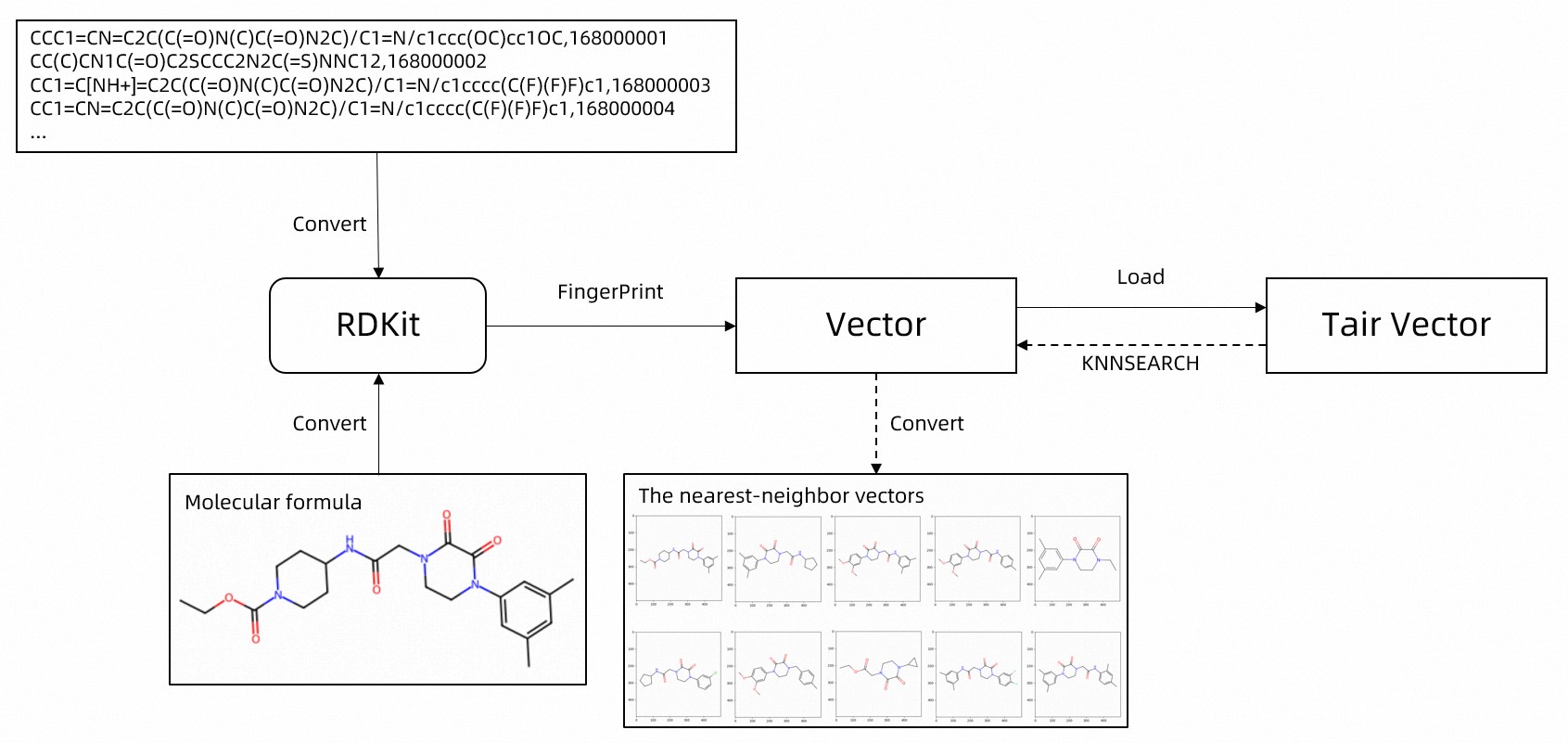
Download the molecular structure dataset.
This example uses test data from the PubChem open source dataset, which contains 11,012 rows of data. The data from the download link is in Simplified Molecular Input Line Entry System (SMILES) format. The following sample shows two columns: the chemical formula and the unique ID.
NoteIn a real-world project, you can write more data to test the millisecond-level retrieval performance of Tair.
CCC1=CN=C2C(C(=O)N(C)C(=O)N2C)/C1=N/c1ccc(OC)cc1OC,168000001 CC(C)CN1C(=O)C2SCCC2N2C(=S)NNC12,168000002 CC1=C[NH+]=C2C(C(=O)N(C)C(=O)N2C)/C1=N/c1cccc(C(F)(F)F)c1,168000003 CC1=CN=C2C(C(=O)N(C)C(=O)N2C)/C1=N/c1cccc(C(F)(F)F)c1,168000004If you download the data from the official website, it is in Structure-Data File (SDF) format. You must convert it to the SMILES format using the following code:
Connect to a Tair instance. For more information, see the
get_tairfunction in the sample code.Create a vector index in Tair to store the molecular structures. For more information, see the
create_indexfunction in the sample code.Write the sample molecular structure data. For more information, see the
do_loadfunction in the sample code.Extract the vector features from the molecular structure data using the RDKit library. Then, use the TVS.HSET vector engine command to store the unique ID, feature information, and chemical formula in Tair.
Perform a similarity search for molecular structures. For more information, see the
do_searchfunction in the sample code.Extract the vector features of the query molecule using the RDKit library. Then, use the TVS.KNNSEARCH vector engine command to query the specified index in Tair for the most similar molecular structures.
Sample code
This example uses Python 3.8. Install the following dependency libraries beforehand: pip install numpy rdkit tair matplotlib.
import os
import sys
from tair import Tair
from tair.tairvector import DistanceMetric
from rdkit.Chem import Draw, AllChem
from rdkit import DataStructs, Chem
from rdkit import RDLogger
from concurrent.futures import ThreadPoolExecutor
RDLogger.DisableLog('rdApp.*')
def get_tair() -> Tair:
"""
Connect to the Tair instance.
* host: The endpoint of the Tair instance.
* port: The port number of the Tair instance. The default is 6379.
* password: The password of the default account for the Tair instance. To connect with a custom account, use the format 'username:password'.
"""
tair: Tair = Tair(
host="r-bp1mlxv3xzv6kf****pd.redis.rds.aliyuncs.com",
port=6379,
db=0,
password="Da******3",
)
return tair
def create_index():
"""
Create a vector index to store molecular structures:
* The index name in this example is "MOLSEARCH_TEST".
* The vector dimension is 512.
* The distance metric is L2.
* The index algorithm is HNSW.
"""
ret = tair.tvs_get_index(INDEX_NAME)
if ret is None:
tair.tvs_create_index(INDEX_NAME, 512, distance_type=DistanceMetric.L2, index_type="HNSW")
print("create index done")
def do_load(file_path):
"""
Enter the path to the molecular structure dataset. This method automatically extracts the vector features of the molecular structures (smiles_to_vector) and writes the data to Tair Vector.
This method also calls the parallel_submit_lines, handle_line, smiles_to_vector, and insert_data functions.
The data is stored in Tair in the following format:
* Vector index name: "MOLSEARCH_TEST".
* Key: The unique ID of the molecular structure, such as "168000001".
* Feature information: 512-dimensional vector information.
* "smiles": The chemical formula, such as "CCC1=CN=C2C(C(=O)N(C)C(=O)N2C)/C1=N/c1ccc(OC)cc1OC".
"""
num = 0
lines = []
with open(file_path, 'r') as f:
for line in f:
if line.find("smiles") >= 0:
continue
lines.append(line)
if len(lines) >= 10:
parallel_submit_lines(lines)
num += len(lines)
lines.clear()
if num % 10000 == 0:
print("load num", num)
if len(lines) > 0:
parallel_submit_lines(lines)
print("load done")
def parallel_submit_lines(lines):
"""
The scheduling method for concurrent writes.
"""
with ThreadPoolExecutor(len(lines)) as t:
for line in lines:
t.submit(handle_line, line=line)
def handle_line(line):
"""
Handles writing a single molecular structure.
"""
if line.find("smiles") >= 0:
return
parts = line.strip().split(',')
try:
ids = parts[1]
smiles = parts[0]
vec = smiles_to_vector(smiles)
insert_data(ids, smiles, vec)
except Exception as result:
print(result)
def smiles_to_vector(smiles):
"""
Extracts the vector features of a molecular structure and converts it from SMILES format to a vector.
"""
mols = Chem.MolFromSmiles(smiles)
fp = AllChem.GetMorganFingerprintAsBitVect(mols, 2, 512 * 8)
hex_fp = DataStructs.BitVectToFPSText(fp)
vec = list(bytearray.fromhex(hex_fp))
return vec
def insert_data(id, smiles, vector):
"""
Writes the vector of a molecular structure to Tair Vector.
"""
attr = {'smiles': smiles}
tair.tvs_hset(INDEX_NAME, id, vector, **attr)
def do_search(search_smiles,k):
"""
Enter the molecular structure to query. This method queries the specified index in Tair and returns the k most similar molecular structures.
First, it extracts the vector features of the query structure. Then, it uses the TVS.KNNSEARCH command to find the unique IDs of the k nearest molecular structures (k=10 in this example). Finally, it uses the TVS.HMGET command to retrieve the corresponding chemical formulas.
"""
vector = smiles_to_vector(search_smiles)
result = tair.tvs_knnsearch(INDEX_NAME, k, vector)
print("The 10 molecular structures most similar to the query target are as follows:")
for key, value in result:
similar_smiles = tair.tvs_hmget(INDEX_NAME, key, "smiles")
print(key, value, similar_smiles)
if __name__ == "__main__":
# Connect to the Tair database and create a vector index for molecular structures named "MOLSEARCH_TEST".
tair = get_tair()
INDEX_NAME = "MOLSEARCH_TEST"
create_index()
# Write the sample data.
do_load("D:\Test\Compound_168000001_168500000.smi")
# In the MOLSEARCH_TEST index, query the 10 molecular structures most similar to "CCOC(=O)N1CCC(NC(=O)CN2CCN(c3cc(C)cc(C)c3)C(=O)C2=O)CC1".
do_search("CCOC(=O)N1CCC(NC(=O)CN2CCN(c3cc(C)cc(C)c3)C(=O)C2=O)CC1",10)The following sample output indicates that the code was executed successfully:
create index done
load num 10000
load done
The 10 molecular structures most similar to the query target are as follows:
b'168000009' 0.0 ['CCOC(=O)N1CCC(NC(=O)CN2CCN(c3cc(C)cc(C)c3)C(=O)C2=O)CC1']
b'168003114' 29534.0 ['Cc1cc(C)cc(N2CCN(CC(=O)NC3CCCC3)C(=O)C2=O)c1']
b'168000210' 60222.0 ['COc1ccc(N2CCN(CC(=O)Nc3cc(C)cc(C)c3)C(=O)C2=O)cc1OC']
b'168001000' 61123.0 ['COc1ccc(N2CCN(CC(=O)Nc3ccc(C)cc3)C(=O)C2=O)cc1OC']
b'168003038' 64524.0 ['CCN1CCN(c2cc(C)cc(C)c2)C(=O)C1=O']
b'168003095' 67591.0 ['O=C(CN1CCN(c2cccc(Cl)c2)C(=O)C1=O)NC1CCCC1']
b'168000396' 70376.0 ['COc1ccc(N2CCN(Cc3ccc(C)cc3)C(=O)C2=O)cc1OC']
b'168002227' 71121.0 ['CCOC(=O)CN1CCN(C2CC2)C(=O)C1=O']
b'168000441' 73197.0 ['Cc1cc(C)cc(NC(=O)CN2CCN(c3ccc(F)c(F)c3)C(=O)C2=O)c1']
b'168000561' 73269.0 ['Cc1cc(C)cc(N2CCN(CC(=O)Nc3ccc(C)cc3C)C(=O)C2=O)c1']Results
You can also plot the similar molecular structures as images. The following figure shows an example.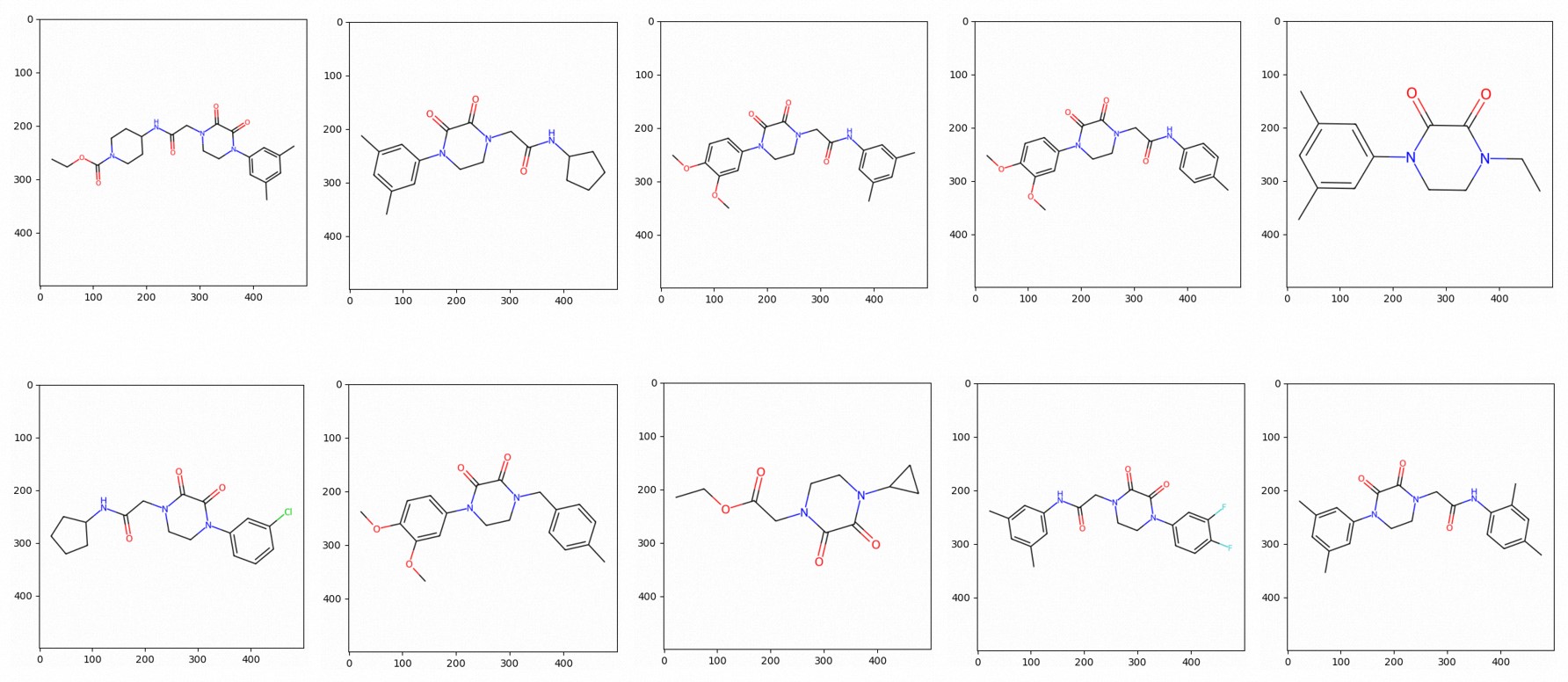
Summary
Using TairVector to search for molecular structures lets you retrieve a list of the most similar structures in milliseconds. As more molecular structure datasets are stored in the Tair database, subsequent queries become more accurate and timely. This solution reduces development time and improves overall efficiency in the field of drug discovery.