このトピックでは、GROMACS を例として使用し、エラスティック・ハイパフォーマンス・コンピューティング(E-HPC)クラスターを使用して分子動力学シミュレーションを実行する方法について説明します。
背景情報
GROningen MAchine for Chemical Simulations(GROMACS)は、完全なソフトウェアパッケージです。数百万個の粒子を含むシステムのニュートン運動方程式をシミュレートすることにより、分子動力学を実行するために使用されます。
GROMACS は、さまざまな複雑な結合相互作用を持つタンパク質や脂質などの生化学分子の核酸分析に使用されます。 GROMACS は、非結合相互作用など、典型的なシミュレーションを計算するための推奨される方法です。さまざまな業界の多くの研究者が、非生物学的システムのポリマーを研究するために GROMACS を使用しています。
GROMACS は、分子動力学(MD)の一般的なアルゴリズムをサポートしています。 グラフィックス・プロセッシング・ユニット(GPU)は、重要な計算タスクを高速化するために GROMACS で使用できます。 詳細については、GROMACS 公式 Web サイトをご覧ください。
関連例
例 1:水中のリゾチーム
この例では、GROMACS を使用して、水箱内のイオンと相互作用するタンパク質をシミュレートします。
この例のファイルをダウンロードするには、次のリンクをクリックします:https://public-ehpc-package.oss-cn-hangzhou.aliyuncs.com/Lysozyme.tar.gz。
例 2:水分子の運動
この例は、水分子のシミュレーションです。 指定された温度で特定の空間内の水分子の動きを追跡するために使用されます。
この例のファイルをダウンロードするには、次のリンクをクリックします:https://public-ehpc-package.oss-cn-hangzhou.aliyuncs.com/water_GMX50_bare.tar.gz。
準備
E-HPC クラスターを作成します。 詳細については、「ウィザードを使用してクラスターを作成する」をご参照ください。
この例では、次のパラメーター構成が使用されます。
パラメーター
説明
ハードウェア設定
デプロイモードを標準に設定します。 2 つの管理ノード、1 つの計算ノード、および 1 つのログオンノードを指定します。 計算ノードとして GPU アクセラレーションインスタンスタイプを選択します。 たとえば、ecs.gn5-c8g1.2xlarge を選択できます。
ソフトウェア設定
CentOS 7.2 パブリックイメージと PBS スケジューラーをデプロイします。 VNC をオンにします。
クラスターユーザーを作成します。 詳細については、「ユーザーを作成する」をご参照ください。
ユーザーは、クラスターへのログオン、ソフトウェアのコンパイル、およびジョブの送信に使用されます。 この例では、次の設定が使用されます。
ユーザー名:gmx.test
ユーザーグループ:sudo 権限グループ
ソフトウェアをインストールします。 詳細については、「ソフトウェアをインストールする」をご参照ください。
次のソフトウェアをインストールします。
Gromacs-gpu V2018.1
openmpi V3.0.0
cuda-toolkit V 9.0
VMD V1.9.3
ステップ 1:クラスターに接続する
次のいずれかの方法を使用してクラスターに接続します。 この例では、ユーザー名として gmx.test を使用します。 クラスターに接続すると、/home/gmx.test に自動的にログオンします。
E-HPC クライアントを使用してクラスターにログオンする
クラスターのスケジューラーは PBS である必要があります。 E-HPC クライアントをダウンロードしてインストールし、クライアントに必要な環境をデプロイしていることを確認してください。 詳細については、「E-HPC クライアントの環境をデプロイする」をご参照ください。
E-HPC クライアントを起動してログオンします。
左側のナビゲーションペインで、[セッション管理] をクリックします。
[セッション管理] ページの右上隅にある [ターミナル] をクリックして、ターミナルウィンドウを開きます。
E-HPC コンソールを使用してクラスターにログオンする
E-HPC コンソール にログオンします。
上部ナビゲーションバーの左上隅で、リージョンを選択します。
左側のナビゲーションペインで、[クラスター] をクリックします。
[クラスター] ページで、クラスターを見つけて [接続] をクリックします。
[接続] パネルで、ユーザー名とパスワードを入力し、[SSH 経由で接続] をクリックします。
ステップ 2:ジョブを送信する
次のコマンドを実行して、ファイルをダウンロードして解凍します。
このトピックでは、水分子の運動の例を使用します。
wget https://public-ehpc-package.oss-cn-hangzhou.aliyuncs.com/water_GMX50_bare.tar.gz tar xzvf water_GMX50_bare.tar.gz chown -R gmx.test water-cut1.0_GMX50_bare chgrp -R users water-cut1.0_GMX50_bare次のコマンドを実行して、gmx.pbs という名前のジョブスクリプトファイルを作成します。
vim gmx.pbsサンプルスクリプト:
説明この例では、gmx.test という名前のユーザーがジョブを送信します。 このジョブは、8 つの vCPU と 1 つの P100 GPU カードを含む計算ノードで実行されます。 計算要件に基づいて計算ノードの構成を調整できます。
#!/bin/sh // ジョブの標準出力と標準エラー出力を同じファイルにリダイレクトします。 #PBS -j oe // 8 つの CPU と 4 つの MPI プロセスを持つ 1 つのノードを選択します。 #PBS -l select=1:ncpus=8:mpiprocs=4 // ジョブを workq キューに送信します。 #PBS -q workq // module コマンドが依存する環境変数を指定します。 export MODULEPATH=/opt/ehpcmodulefiles/ module load gromacs-gpu/2018.1 module load openmpi/3.0.0 module load cuda-toolkit/9.0 export OMP_NUM_THREADS=1 cd /home/gmx.test/water-cut1.0_GMX50_bare/0096 // 例のファイルを前処理して、TPR 入力ファイルを生成します。 /opt/gromacs-gpu/2018.1/bin/gmx_mpi grompp -f pme.mdp -c conf.gro -p topol.top -o topol_pme.tpr // -ntomp は各プロセスに関連付けられた OpenMP スレッドの数を指定し、-nsteps はシミュレーションの反復回数を指定します。 mpirun -np 4 /opt/gromacs-gpu/2018.1/bin/gmx_mpi mdrun -ntomp 1 -nsteps 100000 -pin on -s topol_pme.tpr次のコマンドを実行してジョブを送信します。
qsub gmx.pbs生成されたジョブ ID が 0.scheduler であることを示す次のコマンド出力が返されます。
0.scheduler
ステップ 3:ジョブ結果を表示する
次のコマンドを実行して、ジョブの実行ステータスを表示します。
qstat -x 0.scheduler次のコードは、予期される戻り値の出力の例を示しています。ここで、
S列のRはジョブが実行中であることを示し、S列のFはジョブが完了したことを示します。Job id Name User Time Use S Queue ---------------- ---------------- ---------------- -------- - ----- 0.scheduler gmx.pbs gmx.test 00:34:42 F workq説明ジョブが完了するまで待ちます。 ジョブの完了後、
cat gmx.pbs.o0を実行してジョブの出力を表示できます。VNC を使用してジョブの結果を表示します。
VNC を有効にします。
説明クラスターが属するセキュリティグループで、VNC に必要なポートが有効になっていることを確認してください。 コンソールを使用する場合、システムは自動的にポート 12016 を有効にします。クライアントを使用する場合、ポートを手動で有効にする必要があります。 ポート 12017 では、1 人のユーザーのみが VNC Viewer ウィンドウを開くことができます。 複数のユーザーが VNC Viewer ウィンドウを開く必要がある場合は、ポート 12017 から始まる対応する数のポートを有効にする必要があります。
クライアントを使用する
左側のナビゲーションペインで、[セッション管理] をクリックします。
[セッション管理] ページの右上隅にある [VNC] をクリックして、VNC Viewer を開きます。
コンソールを使用する
E-HPC コンソール の左側のナビゲーションペインで、[クラスター] をクリックします。
[クラスター] ページで、クラスターを選択します。 [詳細] > [VNC] を選択します。
VNC を使用して視覚化サービスにリモート接続します。 詳細については、「VNC を使用して視覚化サービスを管理する」をご参照ください。
クラウドデスクトップの [仮想化サービス] ダイアログボックスで、[アプリケーション] > [システムツール] > [ターミナル] を選択します。
/opt/vmd/1.9.3/vmdを実行して、Visual Molecular Dynamics(VMD)を開きます。VMD メインページで、[ファイル] > [新しい分子...] を選択します。
[参照...] ボタンをクリックし、conf.gro ファイルを選択します。
説明conf.gro ファイルのパスは /home/gmx.test/water-cut1.0_GMX50_bare/0096/conf.gro です。
[読み込み] をクリックします。 [VMD 1.9.3 Opengl ディスプレイ] ウィンドウでジョブの結果を表示できます。

ステップ 4:ジョブのコンピューティングパフォーマンスを表示する
ジョブのコンピューティングパフォーマンスは、コンソールでのみ表示できます。
E-HPC コンソール にログオンします。
左側のナビゲーションペインで、[ジョブとパフォーマンス管理] > [E-HPC チューン] を選択します。
クラスターを見つけて [ノード] をクリックします。
[ノードビュー] タブで、ノードのパフォーマンスを表示します。
ジョブと計算ノードを選択します。
オプション。期間を設定します。
ジョブを選択すると、期間はジョブが実行されている期間に自動的に設定されます。 期間を手動で調整することもできます。
[メトリック] をクリックし、表示するメトリックを選択します。
ノードパフォーマンスデータを表示します。
グラフの上にマウスを置くと、詳細データを表示できます。

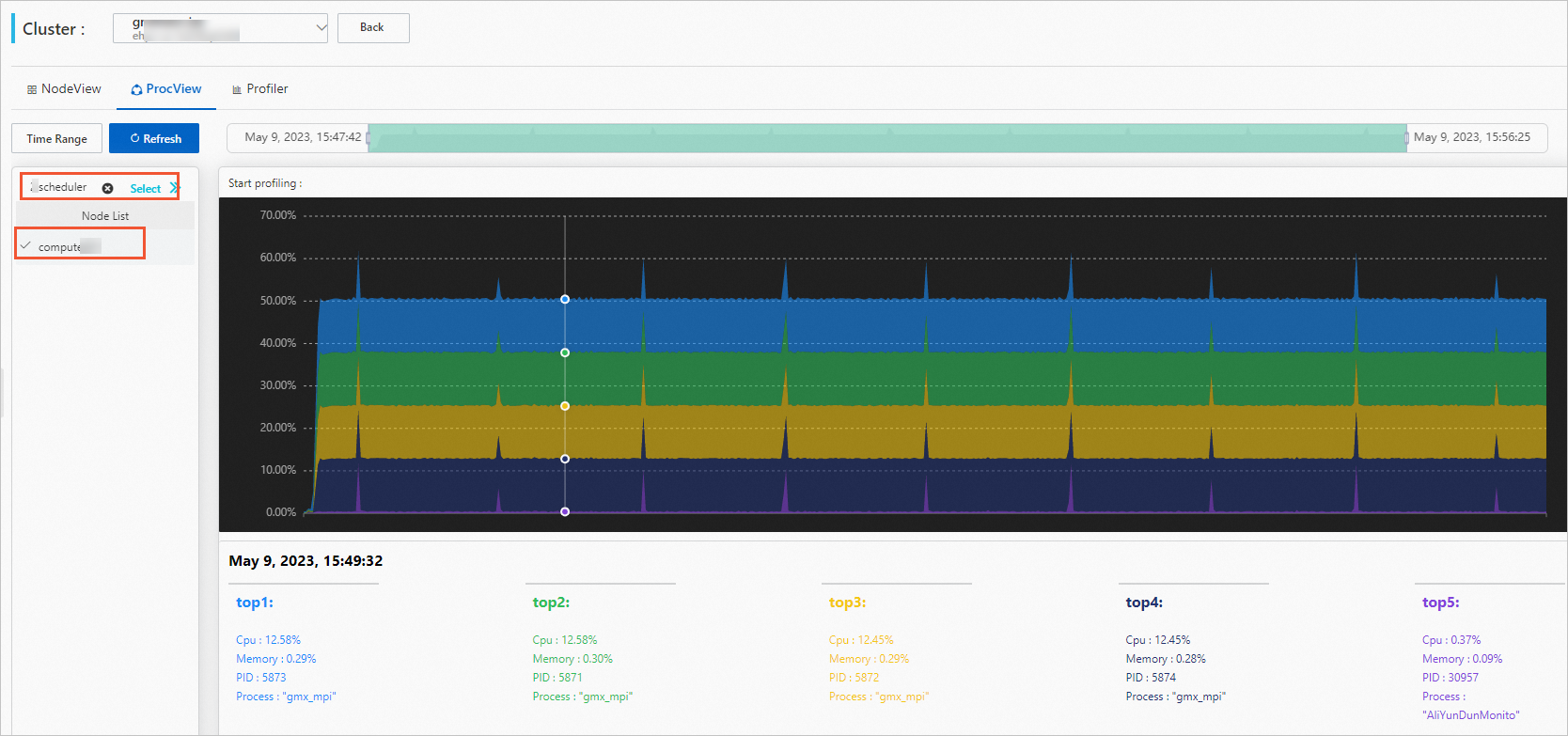
[プロセスビュー] タブをクリックして、プロセスパフォーマンスを表示します。
ジョブと計算ノードを選択します。
オプション。期間を設定します。
ジョブを選択すると、期間はジョブが実行されている期間に自動的に設定されます。 期間を手動で調整することもできます。
プロセスパフォーマンスデータを表示します。
グラフの上にマウスを置くと、詳細データを表示できます。
パフォーマンスタスクのプロファイリングを開始します。
プロセスパフォーマンスグラフで目的の時点をクリックし、グラフの上部でプロファイルするプロセスをクリックします。

表示されたダイアログボックスで、プロファイリングパラメーターを構成します。
プロファイリングの期間と頻度を設定し、[OK] をクリックしてジョブのリアルタイムプロファイリングを開始します。
[プロファイラー] タブをクリックして、プロファイリング結果を表示します。
プロファイリングタスクを選択し、[表示] をクリックします。
プロファイリング結果を表示します。
グラフの上にマウスを置くと、詳細データを表示できます。
